Von der Evidenz in die Versorgung: Chancen und Risiken für Gesundheits- und Wissenschaftspolitik. Ein Positionspapier der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF)
Bernhard Wörmann 1Ina Kopp 2
Monika Nothacker 2
Ernst Klar 3
Martin Sedlmayr 4
Michael Vogeser 5
Henning Schliephake 6
Rolf-Detlef Treede 7,8
1 Medizinische Klinik mit Schwerpunkt Hämatologie, Onkologie und Tumorimmunologie, Charité – Universitätsmedizin Berlin, Deutschland
2 AWMF-Institut für medizinisches Wissensmanagement (AWMF-IMWi), Marburg/Berlin, Deutschland
3 AWMF-Ad-hoc-Kommission Bewertung von Medizinprodukten, Berlin, Deutschland
4 Institut für Medizinische Informatik und Biometrie, Medizinische Fakultät Carl Gustav Carus, TU Dresden, Deutschland
5 Institut für Laboratoriumsmedizin, Klinikum der Universität München, Ludwigs-Maximilian-Universität (LMU) München, Deutschland
6 Klinik für Mund-, Kiefer- und Gesichtschirurgie, Georg-August-Universität Göttingen, Deutschland
7 Abteilung für Neurophysiologie, Mannheimer Centrum für Translationale Neurowissenschaften, Universität Heidelberg, Mannheim, Deutschland
8 Abteilung für Psychiatrie und Psychotherapie, Zentralinstitut für Seelische Gesundheit, Medizinische Fakultät Mannheim, Universität Heidelberg, Mannheim, Deutschland
Zusammenfassung
Fragestellung: Nach über 25 Jahren der Leitlinienentwicklung hat die Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) in einem Symposium die folgenden Themen zur Verbesserung der Implementierung von Evidenz in die Versorgung diskutiert: Verbreiterung der Datenbasis für die Leitlinienentwicklung, regulatorische Rahmenbedingungen für diese Erweiterung, Transfer der Leitlinienaussagen in die medizinische Praxis sowie Chancen und Risiken, die sich hierfür aus der europäischen Gesetzgebung ergeben.
Methoden: Auf dem Berliner Forum der AWMF am 27.04.2022 trugen Fachleute aus wissenschaftlichen Fachgesellschaften und nationalen Institutionen des Gesundheitswesens ihre Erfahrungen und Vorstellungen zu den o.g. Themen vor. Hieraus und aus den Diskussionsbeiträgen beim Berliner Forum trugen drei Schreibgruppen die wesentlichen Aussagen für diesen Artikel zusammen.
Ergebnisse: Die AWMF empfiehlt:
– Schaffung einer digitalen Infrastruktur für die Qualitätssicherung von Leitlinien und für die Verfügbarkeit von deren Inhalten am Krankenbett und bei der Konsultation
– Ausweitung industrieunabhängiger klinischer Studien zu Prävention, Diagnostik und Therapie mit Medikamenten, Medizinprodukten oder anderen Verfahren
– Förderung von Registerstrukturen zur Erzeugung von versorgungsnahen Daten in der Gesundheitsversorgung
– Reduktion übertriebener bürokratischer Hürden auf nationaler und EU-Ebene
Schlussfolgerungen: Mit den konkreten Empfehlungen dieses Positionspapiers weist die AWMF auf notwendige Schritte zur Verbesserung der Translation von der Evidenz in die klinische Versorgung hin.
Schlüsselwörter
Leitlinien für die klinische Praxis, Versorgung, evidenzbasierte Medizin, Medizinprodukteverordnung, Point-of-Care, Leitlinienregister
1 Einleitung
Auf Empfehlung des Sachverständigenrats für die Konzertierte Aktion im Gesundheitswesen koordiniert die AWMF (Arbeitsgemeinschaft Wissenschaftlicher Medizinischer Fachgesellschaften) seit 1996 die Erstellung und Aktualisierung von medizinischen Leitlinien für Deutschland [1]. Ihre Mitglieder leisten damit einen wesentlichen Beitrag für die Implementierung einer evidenzbasierten Medizin. Die Prozesse der Leitlinienentwicklung und deren Publikation für Fachpublikum und Laien sind inzwischen auf hohem Niveau standardisiert, und mittels dieser Methodik kann die AWMF der Gesundheitspolitik fundierte Fakten als Basis für Entscheidungsfindung effizient und umfassend liefern. Diese Zuarbeit hat sich u.a. in der COVID-19-Pandemie bewährt.
Die AWMF hat politische Forderungen zur Nutzung der evidenzbasierten Medizin als Grundlage der Gesundheitspolitik veröffentlicht (Tabelle 1 [Tab. 1]), [2], [3]. Die Herausarbeitung konkreter Handlungsanweisungen zur optimalen Generierung von hochwertigen Gesundheitsdaten und deren praktischer Umsetzung in der Krankenversorgung waren Thema eines Berliner Forums der AWMF. Kernpunkt der Diskussion mit Sachverständigen aus Medizin und Gesundheitspolitik war die nachhaltige Förderung von Leitlinien als Grundlage einer wissenschaftlich informierten Gesundheitspolitik. Hierzu gehört die Verbesserung der Rahmenbedingungen für Gesundheitsforschung und Forschungstransfer in Deutschland [4] und die vorausschauende Begleitung der Rahmenbedingungen und Entwicklungen im europäischen Kontext.
Tabelle 1: Politische Forderungen der AWMF
Um neue Erkenntnisse der wissenschaftlichen Medizin zügig in die Krankenversorgung einzubringen, ist ein guter Informationsfluss von den wissenschaftlichen Akteuren zu den politisch Entscheidenden nötig. Diese von der AWMF erstmals zur Bundestagswahl 2017 erhobene Forderung wird zunehmend in die Praxis umgesetzt. Zu allen fünf Themenbereichen gibt es aber auch in der aktuellen Legislaturperiode weiteren Handlungsbedarf.
Quelle: AWMF [21], [22]
2 Methodik
Im Rahmen des Berliner Forums der AWMF vom 27. April 2022 wurde von Sachverständigen aus den Mitgliedsgesellschaften der AWMF und aus Institutionen des Gesundheitswesens zum Status Quo und zu Stärken und Schwächen der nationalen und europäischen Rahmenbedingungen der Gesundheitsforschung und deren Bedeutung für die Translation von Leitlinien in die klinische Praxis referiert und mit Vertretern relevanter wissenschaftlicher medizinischen Fachgesellschaften und weiterer Organisationen im Gesundheitswesen diskutiert. Die Inhalte und die abgeleiteten Empfehlungen wurden durch drei Schreibgruppen in einen Textentwurf gefasst. Dieser wurde zwischen den beteiligten Referierenden mehrfach abgestimmt, eingehend im AWMF-Präsidium diskutiert und in der vorliegenden finalen Fassung konsentiert (Tabelle 2 [Tab. 2]).
Tabelle 2: Vorträge beim Berliner Forum der AWMF vom 27. April 2022
Quelle: AWMF [23]
3 Rahmenbedingungen für Gesundheitsforschung und Forschungstransfer in Deutschland
Eine entscheidende Voraussetzung für den Transfer von Forschungsergebnissen in die medizinische Versorgung ist die Existenz bzw. die Schaffung von geeigneten Rahmenbedingungen für die unabhängige Durchführung von klinischen Studien. Dies war das erste der drei Schwerpunktgebiete im Rahmen des Berliner Forums 2022, diskutiert unter dem Vorsitz von Prof. Dr. Rolf-Detlef Treede.
3.1 Arzneimittelversorgung
Eine zentrale Rolle hat das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) an der Schnittstelle zwischen europäischer Zulassung und nationaler Versorgung. Aktuelle Schwerpunkte und Herausforderungen sind:
- Klinische Prüfungen – Aktivitäten für den Standort Europa: Basis der Förderung qualitativ hochwertiger, klinischer Prüfungen ist die EU Clinical Trials Regulation aus dem Jahr 2014, umgesetzt seit dem Januar 2022 [5]. Sie wird von den nationalen HMA (Heads of Medicines Agencies) und der europäischen Institution EMA (European Medicines Agency) in der Netzwerkstrategie 2025 weiterentwickelt. Dabei werden die grundlegenden Prinzipien der „Good Clinical Practice“ (GCP) beibehalten, aber stärker am Bedarf orientiert. Unnötiger bürokratischer Aufwand soll vermieden werden; Antragsverfahren sollen vereinfacht und transparenter werden.
- Unterstützungen seitens der Zulassungsbehörden: Durch die Initiative ACT EU soll die EU als Drehscheibe für die klinische Forschung weiter etabliert, die Entwicklung hochwertiger, sicherer und wirksamer Arzneimittel weiter gefördert und gleichzeitig die klinische Forschung besser in das europäische Gesundheitssystem integriert werden. Zentrale Elemente sind der rasche Ausbau funktionsfähiger, digitaler Plattformen sowie des Programms STARS (Strengthening Training of Academia in Regulatory Science) zur Fort- und Weiterbildung von klinischen Wissenschaftlern (Abbildung 1 [Abb. 1]).
- Regulatorisches Umfeld – „lessons learned“ aus der Pandemie für ein zukunftsfähiges regulatorisches System: COVID-19 hat auch die EMA gefordert und Änderungen bewirkt. Zentrale Botschaft aus der Pandemie ist die Notwendigkeit einer maximalen Flexibilität, Agilität und Kooperation. Durch das Verfahren des Rolling Review konnte eine schnellere Zulassung u.a. für Impfstoffe erreicht werden. Nachteile sind die größere Unsicherheit der Daten, weniger Zeit für die Bewertung der Daten und die Verlagerung der Bewertung wichtiger Daten und Analysen in die Zeit nach der Zulassung. Ein positiver Nebeneffekt der Pandemie war der „digitale Push“ für versorgungsnahe Daten.
Abbildung 1: Rahmenbedingungen und Entwicklungen im europäischen Kontext
Das Projekt ACT-EU dient der Beschleunigung von klinischen Studien in der Europäischen Union. Die dort genannten Elemente sollten auch in die deutsche Gesetzgebung einfließen. Der ACT EU 2022–2026 Arbeitsplan enthält die geplanten Projektergebnisse und Zeitabläufe für den Zeitraum 2022–2026. Für 2023 sind unter anderem folgende Projektergebnisse geplant: die Etablierung eines Prozesses zur Unterstützung akademischer Sponsoren bei der Durchführung großer multinationaler klinischer Studien, das Aufsetzen einer Plattform für den Dialog zwischen den verschiedenen an klinischen Studien beteiligten Interessengruppen unter Einbeziehung von Patientinnen und Patienten, Angehörigen von Gesundheitsberufen und Mitwirkenden aus dem akademischen Bereich sowie die Förderung von Innovationen in der Methodik dezentraler klinischer Studien.
Abbildung zusammengesetzt aus Kopfzeile und Titel von [24] (Copyright: Heads of Medicines Agencies, European Commission und European Medicines Agency) und der Grafik „ACT EU“ aus [25] (Copyright: European Medicines Agency)
Zentrale Botschaft des Vortrags war die Notwendigkeit eines innovationsfreundlichen „Mindset“ für moderne Patientenversorgung.
3.2 Nutzenbewertung von Arzneimitteln
Eine wichtige Funktion bei der Integration neuer Arzneimittel in die Versorgung haben die wissenschaftlichen medizinischen Fachgesellschaften. Ihr inhaltliches Spektrum reicht von der Grundlagenforschung, der Durchführung klinischer Studien, der Erstellung von Leitlinien bis zur Bewertung des Zusatznutzens von Arzneimitteln. Schwerpunkte des Vortrags waren:
- Beteiligung der Fachgesellschaften am Prozess der frühen Nutzenbewertung neuer Arzneimittel (AMNOG-Verfahren): Das AMNOG-Verfahren dient der Preisbildung und ist primär keine Kernaufgabe der Fachgesellschaften. Ihre Aufgabe ist aber die Sicherung der evidenzbasierten Bewertung aus klinisch-wissenschaftlicher Sicht und damit die Brücke zwischen den Bewertungen des Gemeinsamen Bundesausschusses (G-BA) und den Leitlinien der Fachgesellschaften [6]. Die Fachgesellschaften haben sich in den letzten Jahren an etwa 90% aller Bewertungsverfahren beteiligt [7]. Ihre zunehmende Rolle wurde durch eine Gesetzesänderung durch das Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) aus dem Jahr 2019 anerkannt. Danach werden die Fachgesellschaften jetzt auch in die frühe Beratung des G-BA durch Erstellung gutachterlicher Expertisen einbezogen.
- Methodische Defizite des Bewertungsverfahrens: In zentralen methodischen Fragen unterscheidet sich das AMNOG-Verfahren von der Methodik der Leitlinien. Hier stehen Konzepte des Health Technology Assessment mit Fokus auf Zusatznutzen und Kosteneffizienz den Konzepten der an individuellen Patienten und Patientinnen ausgerichteten evidenzbasierten Medizin gegenüber. Das kann zu Diskrepanzen bei den Bewertungen und nachfolgend auch zu Unsicherheit in der Versorgung, konkret in der Verschreibung von Arzneimitteln, führen. Unterschiedliche Methodik gibt es bei der Bildung von Subgruppen, aber auch bei inhaltlichen Aspekten der Bewertung von Endpunkten wie der Verlängerung der Gesamtüberlebenszeit, der Verlängerung von krankheitsfreiem oder progressionsfreiem Überleben, Nebenwirkungen, Lebensqualität und weiteren Parametern des Patient-Reported-Outcome.
- Arzneimittelengpässe: Was helfen die besten Konzepte und Bewertungen, wenn ein Arzneimittel aufgrund eines Lieferengpasses nicht zur Verfügung steht? Auch hier haben die Fachgesellschaften in den letzten Jahren eine zentrale Rolle übernommen. In Abhängigkeit ihrer Bewertung, ob ein Arzneimittel unverzichtbar ist oder ob es äquieffektive Alternativen gibt, greifen die neueren gesetzlichen Regelungen für den erleichterten Import von Arzneimitteln aus dem Ausland.
Zusammenfassend ist es unerlässlich, dass die zunehmende Anerkennung der Rolle von Fachgesellschaften von diesen auch angenommen werden muss. Dazu gehören Professionalisierung, Optimierung organisatorischer Strukturen und bei einigen Fachgesellschaften auch eine Änderung des „Mindset“ von der eigenen Profilierung zu Kooperation.
3.3 Klinische Studien
Die Koordinierungszentren Klinische Studien (KKS) haben in den letzten Jahrzehnten in Deutschland eine zentrale Rolle bei der inhaltlichen und organisatorischen Gestaltung klinischer Studien übernommen [8]:
- Geschichte: Bereits vor über 20 Jahren wurde in Deutschland ein Mangel an patientenorientierter, klinischer Forschung diagnostiziert. Das KKS-Netzwerk entstand nach zwei Ausschreibungsrunden des Bundesministeriums für Bildung und Forschung (BMBF). Ziel war die Schaffung studienbezogener Wissenschaftseinrichtungen an universitätsmedizinischen Standorten, ausgerichtet an internationalen Standards.
- Leistungen: Die Zahl der Mitglieder ist seit der Gründung kontinuierlich gestiegen. Im Jahr 2022 sind 25 universitätsmedizinische Standorte Mitglied im KKS e. V. Bei einer Auswertung aus dem Jahr 2020 hatten die Mitglieder des KKS den zentralen Support von 1.203 Studien übernommen. Unterstützt werden sowohl Investigator Initiated Trials (IIT) als auch Industry Sponsored Trials (IST). Zentrale Aufgabe des KKS-Netzwerkes ist dabei neben der praktischen Durchführung auch die Förderung der Fort- und Weiterbildung an den Standorten. Im Jahr 2020 wurden 319 Grundlagen-, Aufbau-, Refresher- und Spezialkurse mit mehr als 7.800 Teilnehmenden an den Standorten durchgeführt.
- Ziele erreicht? Erreicht wurden eine Steigerung der Qualität klinischer Studien, die Etablierung der Clinician Scientist Programme, die Förderprogramme für klinische Studien seitens der Deutschen Forschungsgemeinschaft (DFG) und des BMBF sowie eine zunehmende Evidenzorientierung der Politik. Auf der Negativseite stehen zu wenige IITs im Vergleich zu anderen Staaten, keine Auslastung der Förderprogramme, das Fehlen zentraler Studienstrategien, eine Heterogenität der Standorte, ökonomischer Druck in der Universitätsmedizin und weitere Barrieren in organisatorischen und inhaltlichen Belangen.
Zusammenfassend ist zu betonen, dass qualitativ hochwertige klinische Studien zu den grundlegenden Bausteinen einer Evidenz-Landschaft gehören, von denen das gesamte Gesundheitssystem profitiert. Voraussetzungen sind interdisziplinäres Teamwork, gute Netzwerke und der Willen zu einer evidenzbasierten Gesundheits- und Forschungspolitik (Abbildung 2 [Abb. 2]).
Abbildung 2: Prototypische Abfolge der Planung und Durchführung einer klinischen Studie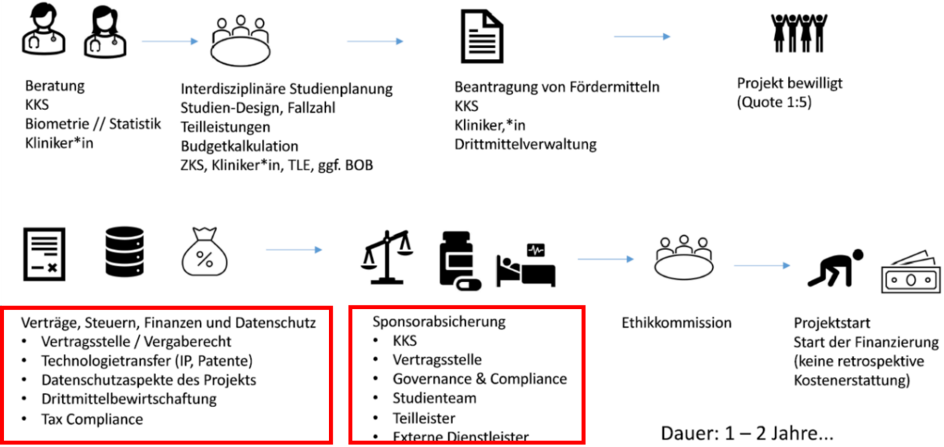
Klinische Studien an und mit kranken oder gesunden Menschen bilden die Grundpfeiler der medizinischen Versorgung im Sinne einer evidenzbasierten Medizin, die sich in ständigem Wandel befindet. Dies gilt einerseits für die Zulassung von Arzneimitteln, Medizinprodukten oder digitalen Gesundheitsanwendungen, andererseits auch für die Erstellung und Aktualisierung medizinischer Leitlinien für die klinische Praxis. Dieses Schema illustriert die Komplexität der Planung und Durchführung solcher Studien. Die roten Kästen markieren zeitaufwändige Prozesse, die von Bürokratieabbau profitieren würden.
Legende: BOB=Bundesoberbehörde IP=intellectual property; KKS=Koordinierungszentrum für Klinische Studien, ZKS=Zentrum für Klinische Studien Tübingen, TLE=Trial Evaluation Center
Quelle: Vortrag B. Lang beim Berliner Forum, Netzwerk KKS (https://www.kks-netzwerk.de/netzwerk/ueber-uns/)
3.4 Empfehlungen
Für einen besseren Transfer von Evidenz in die Versorgung empfiehlt die AWMF:
- Enge Kooperation der nationalen mit der EU-Ebene
- Durchgehende Integration evidenzbasierter Medizin in klinische Studien von der Planung über die Durchführung bis zu Früh- und Langzeitauswertungen
- Bei Studien, die von der pharmazeutischen Industrie initiiert und finanziert werden: Beteiligung der Fachgesellschaften an den Konsultationen der regulatorischen Behörden zur Sicherstellung des Bezugs neuer Arzneimittelzulassungen zum aktuellen Stand der Versorgung
- Bei allen klinischen Studien in Deutschland: Nutzung der Ressourcen der professionellen Koordinierungszentren klinischer Studien (KKS) zur Sicherung der Studienqualität und zur Steigerung der Zahl eingeschlossener Patientinnen und Patienten
- Abbau überbordender bürokratischer Hürden, auch beim Umsetzen von Datenschutzregelungen
- Förderung unabhängiger klinischer Studien in Form von IITs (Investigator initiated Trials) und in Form von Versorgungsstudien
- Schaffung der gesetzlichen Möglichkeit für Kostenträger (Krankenkassen) zur aktiven Beteiligung an klinischen Studien
4 Leitlinien als Grundlage einer wissenschaftlich informierten Gesundheitspolitik
Eine weitere Voraussetzung für den Transfer von Forschungsergebnissen in die medizinische Versorgung ist die Erstellung und regelmäßige Aktualisierung von qualitativ hochwertigen Leitlinien sowie die schnelle Verfügbarkeit der Leitlinienaussagen am Krankenbett. Dies war das zweite Schwerpunktgebiet des Berliner Forums 2022, diskutiert unter dem Vorsitz von Prof. Dr. Ina Kopp.
4.1 Umsetzung der Leitlinienarbeit durch die AWMF und ihre Fachgesellschaften
Die Verantwortung für Leitlinien liegt in Deutschland bei den wissenschaftlichen medizinischen Fachgesellschaften. Die AWMF koordiniert und unterstützt die Initiativen der Fachgesellschaften seit 1995 [1], [9], indem sie ein qualitätsgesichertes Leitlinienregister aufgebaut hat sowie methodische Beratung durch das AMWF-Institut für Medizinisches Wissensmanagement zur Verfügung stellt [10]. Alle Leitlinienprojekte werden angemeldet, hinsichtlich ihrer Entwicklungsmethodik (S1–S3) klassifiziert und bei Einreichung formal auf die Einhaltung der Anforderungen überprüft, inklusive eines obligaten Interessenkonfliktmanagements. Das zugrundeliegende AWMF-Regelwerk wird von der Ständigen Kommission Leitlinien der AWMF regelhaft angepasst [11]. Mit hohem Engagement der Fachgesellschaften hat sich diese Vorgehensweise auch während der COVID-19-Pandemie bewährt. Stand April 2022 befanden sich 810 publizierte Leitlinien im AWMF-Register, davon 17 zu COVID-19 [11], [12]. Die Leitlinien-Task-Force der AWMF arbeitete dabei zusammen mit dem Netzwerk Universitätsmedizin [13].
Die Qualität der Leitlinien hat sich kontinuierlich verbessert; immer mehr Leitlinien weisen mittlerweile die höchste methodische Klasse (S3) auf. Aufgrund der gestiegenen methodischen Anforderungen, z.B. für Evidenzrecherchen und Dokumentation, und der höheren Zahl an Leitlinien haben sich viele Fachgesellschaften für die Leitlinien-Arbeit professionalisiert und unterstützen diese durch eigene Leitliniensekretariate sowie ehrenamtliche Leitlinienkommissionen oder Leitlinienbeauftragte.
4.2 Nutzung von Leitlinien durch den Gemeinsamen Bundesausschuss
Leitlinien werden von klinisch Tätigen genutzt, aber auch von Institutionen im Gesundheitswesen. Der Gemeinsame Bundesausschuss (G-BA) recherchiert regelhaft Leitlinienempfehlungen für die Ermittlung des Versorgungsstandards, beispielsweise für Disease-Management-Programme (DMP), Qualitätssicherungsmaßnahmen, zweckmäßige Vergleichsinterventionen/-therapien für neue Arzneimittel im Rahmen der Frühen Nutzenbewertung und für nicht-medikamentöse Methoden sowie Erprobungsrichtlinien. Ebenso spielen Leitlinien eine Rolle, um Versorgungsbereiche in verschiedenen Regelungskontexten zu strukturieren (z.B. in der Psychotherapie für die „Personalausstattung in der Psychiatrie und die Psychosomatik-Richtlinie“) oder um Übersichten zu Präventions- und Therapieempfehlungen in anderen Ländern zu stellen (z.B. nicht-invasiver Pränataltests). Der G-BA erwartet systematisch erstellte, evidenzbasierte, methodisch hochwertige Leitlinien, in denen eine Nutzung bzw. Auseinandersetzung mit vorhandenen öffentlichen Informationsquellen (z.B. G-BA, IQWiG) erfolgt und die Aussagen zu patientenrelevanten Endpunkten enthalten. Besonderer Wert wird auf Unabhängigkeit von der Industrie gelegt sowie auf Transparenz bezüglich Finanzierung und potenzieller Interessenskonflikte der Leitliniengruppenmitglieder. Außerdem müssen Leitlinien auf den deutschen Versorgungskontext übertragbar und die Begründung für die Ableitung der Empfehlungen nachvollziehbar sein. Im G-BA werden zu verschiedenen Themen hochwertige Leitlinien vermisst, wie z.B. zu Fettstoffwechselstörungen bei Erwachsenen oder zur Betreuung von Schwangeren. Für andere Themen wie beispielsweise Neurodermitis oder Migräne liegen zwar Leitlinien vor, aber keine evidenz- und konsensbasierten S3-Leitlinien.
4.3 Unterstützung der Leitlinienentwicklung durch öffentliche Mittel
Lange Zeit wurden Leitlinien nicht öffentlich finanziert. Mit dem In-Kraft-Treten des Digitale-Versorgung-Gesetzes 2019 hat sich dies geändert. Aus den Mitteln des Innovationsfonds für die Versorgungsforschung stehen bis 2024 jährlich mind. 5 Millionen Euro für Leitlinienprojekte zur Verfügung (§ 92b Absatz 2 SGB V) und das Institut für Qualität und Wirtschaftlichkeit (IQWiG) unterstützt nun Leitlinienprojekte mit Evidenzrecherchen zu einzelnen Fragestellungen mit bis zu 2 Millionen Euro pro Jahr (§ 139b). Anträge können für neue Leitlinien, aber auch für Aktualisierungen gestellt werden. Auf die Ausschreibungen 2020/2021 gingen 62 Anträge ein, von denen 41 mit insgesamt 12,7 Millionen Euro gefördert werden [14]. Gebunden sind die Einreichungen allerdings an Themenschwerpunkte, die das Bundesministerium für Gesundheit (BMG) festlegt. Die AWMF reicht hierzu regelmäßig in der Leitlinienkommission abgestimmte Vorschläge ein und setzt sich darüber hinaus für eine themenoffene Förderung ein. Die Erstellung von Evidenzberichten durch das IQWiG befindet sich in der Erprobung; erst wenige Berichte sind fertiggestellt. Die Fragestellungen decken jeweils nur einen kleinen Teil des Gesamtspektrums der jeweiligen Leitlinienempfehlungen ab.
Die AWMF begrüßt die neuen Finanzierungsmöglichkeiten für Leitlinienprojekte. Die bereitgestellten Mittel decken nur einen geringen Teil der Kosten ab, die den Beteiligten der derzeit über 100 angemeldeten S3-Leitlinien entstehen. Sie sind aber ein wichtiger Anreiz, sich für methodisch hochwertige Leitlinien zu engagieren.
4.4 Digitalisierung des Leitlinienwissens
Um durch Leitlinien zukunftsgerichtet Evidenz in die Versorgung zu bringen, besteht nach Einschätzung der AWMF die Notwendigkeit der Digitalisierung der Leitlinien sowie des AWMF-Leitlinienregisters [15]. Damit Leitlinien vor Ort genutzt werden können, sind zudem digitale Schnittstellen zum jeweiligen „Point of Care“ erforderlich, damit Leitlinienempfehlungen in geeigneter Form die gemeinsame Entscheidungsfindung unterstützen können. Um die Digitalisierung voranzutreiben, wurden seitens des AWMF-Instituts für Medizinisches Wissensmanagement Pilotprojekte durchgeführt [16]. Viele Fachgesellschaften sind in Bezug auf Digitalisierung aktiv und benötigen strukturelle Unterstützung. Neben der Implementierung kann auch die Evaluation der Leitlinienumsetzung mit digitaler Unterstützung vereinfacht werden [17]. Evaluationen zur Überprüfung des Nutzens von Leitlinien und zum Aufzeigen von Forschungslücken sind unabdingbar und sollten Grundlage jeder Aktualisierung sein (Abbildung 3 [Abb. 3]).
Abbildung 3: Leitlinien als Grundlage einer wissenschaftlich informierten Gesundheitspolitik 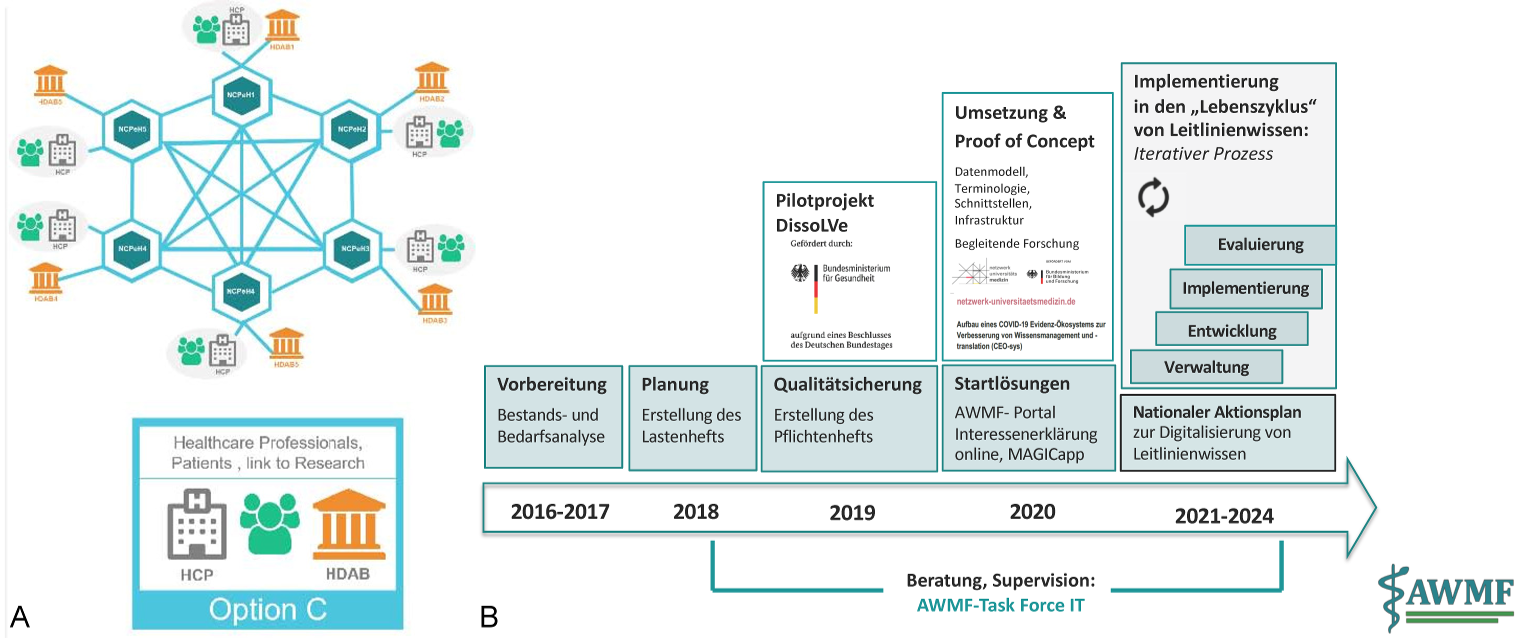
A: Das Konzept des Europäischen Gesundheitsdatenraums (European Health Data Space EHDS): zielt auf die Integration grenzüberschreitender professioneller Gesundheitsdienstleistungen (e-Rezept, ePatientenakte), bürgerzentrierter Dienstleistungen (mobile Apps zur Speicherung und Zugangskontrolle für eigene Gesundheitsdaten), anderer Dienstleistungen (Telemedizin, Interoperabilität der Infrastrukturen, Teilen von Patientendaten auch jenseits von EU/EEA), und den Zugang zur sekundären Nutzung von Gesundheitsdaten durch legitime Interessengruppen (Forschende, Entscheidungstragende etc.). Copyright Abbildung 3A: European Commission [26]
B: Das Leitlinienprojekt der AWMF bereitet die deutschen Leitlinien auf die Einbindung in den deutschen und europäischen digitalen Gesundheitsdatenraum vor. Copyright Abbildung 3B: AWMF [15]
4.5 Empfehlungen
Für eine bessere Nutzung von Evidenz in der Versorgung empfiehlt die AWMF:
- die Integration der umfassenden Digitalisierung von Leitlinien in die Digitalisierungsstrategie des Bundesministeriums für Gesundheit mit dem Ziel, nachhaltige digitale Strukturen zu der Qualitätssicherung von Leitlinien einerseits und der Wissensvermittlung an den „Point of Care“ andererseits zu ermöglichen. Neben der integrierten Konzeption und Finanzierung erfordert dies auch gesetzliche Regelungen für einheitliche ambulante und stationäre Schnittstellen.
- das Weiterführen der Leitlinienprojekt-Finanzierung im Rahmen des Innovationsfonds nach 2024 mit der Möglichkeit auch der themenoffenen Förderung wie für Versorgungsforschungsprojekte.
- die gezielte Förderung von Versorgungsforschungsprojekten zur Implementierung und Evaluierung von methodisch hochwertigen Leitlinien (S3).
5 Rahmenbedingungen und Entwicklungen im europäischen Kontext
In den kommenden Jahren muss sich das Gesundheitswesen mit Entwicklungen auf europäischer Ebene auseinandersetzen, die die klinische Versorgung im nationalen Rahmen in erheblichem Maße beeinflussen werden. Dies betrifft einerseits Regelungen, die sich auf die Aufrechterhaltung und Neuentwicklung diagnostischer und therapeutischer Prozeduren beziehen, und andererseits Entwicklungen im IT-Bereich, die die integrative Nutzung großer Datenmengen zur Verbesserung der medizinischen Versorgung ermöglichen sollen. Dies war das dritte Schwerpunktgebiet des Berliner Forums 2022 unter Vorsitz von Prof. Dr. Dr. Henning Schliephake.
5.1 Medical Device Regulation (MDR)
Zu den europäischen Regelungen mit erheblichem Einfluss auf die Gesundheitsversorgung gehört die Medical Device Regulation (MDR), die 2017 von den europäischen Gremien verabschiedet wurde und sich seit Mai 2021 in der Umsetzung auf nationaler Ebene befindet. Ziel der MDR ist es, u.a. eine verbesserte Transparenz auf Basis der Produktidentifizierungsnummer (Unique Device Identification/UDI) und der Europäischen Datenbank für Medizinprodukte (EUDAMED-Datenbank) durch automatisierbare Identifizierung von Produkten zu garantieren. Darüber hinaus soll mit der MDR durch die Implementierung neuartiger Lebenszyklusprozesse zur klinischen Bewertung und zum Risikomanagement der Medizinprodukte sowie zur proaktiven Marktbeobachtung durch den Hersteller eine Erhöhung der Patientensicherheit erreicht werden. Bei der Implementierung der Neuregelung in die Versorgungsrealität auf nationaler Ebene müssen jedoch eine Reihe von Hürden überwunden werden.
Die drei größten Herausforderungen bei der Umsetzung der MDR sind aus heutiger Sicht
- die geforderten klinischen Prüfungen für die große Zahl der Bestandsprodukte,
- die erheblichen Kostensteigerungen für die Rezertifizierung und
- die fehlenden Kapazitäten bei den Benannten Stellen.
Die MDR stellt prinzipiell für Bestandsprodukte keine unerfüllbaren neuen oder geänderten Anforderungen an die Marktfähigkeit von Medizinprodukten und enthält Bestimmungen, mit denen eine weitere Verfügbarkeit der Produkte gewährleistet werden kann, sofern der Hersteller nachweisen kann, dass er die notwendigen Schritte rechtzeitig eingeleitet hat. Für diesen Prozess bestehen für Bestandsprodukte Übergangsfristen von 3–5 Jahren, die aus Sicht des Bundesministeriums für Gesundheit ausreichen sollten, um klinische Daten zur Rezertifizierung von Bestandsprodukten vorzulegen. Allerdings wurden bisher nur für 10% aller anstehenden Bestandsprodukte Anträge auf Rezertifizierung gestellt. Hier ist eine Zurückhaltung der Hersteller erkennbar, die umso mehr erstaunt, da der Bestandsschutz unter den neuen Vorgaben eingeschränkt ist. Einer der Gründe für diese Zurückhaltung ist die von den Herstellern beklagte hohe Belastung sowohl in finanzieller Hinsicht als auch in der Bindung von Manpower für die MDR-regulierte Rezertifizierung von Bestandsprodukten. Der niedrige Stand der bisher durchgeführten Rezertifizierungen kann daher ein Vorzeichen für eine möglicherweise drohende Ausdünnung des Marktes an Bestandsprodukten in den folgenden Jahren sein. Das Problem der hohen administrativen Hürden verschärft sich noch für den Bereich der Nischenprodukte (Orphan Devices), da hier die Erbringung eines vollständigen klinischen Nachweises ein offensichtliches Problem darstellt. Im Hinblick auf die dritte Hürde bei der Umsetzung der MDR, die Kapazität der Benannten Stellen, beginnt sich der bestehende Engpass durch zu wenige Benannte Stellen aufzulösen. Bis Ende 2023 wird mit einer Steigerung der Zahl auf 44 gerechnet. Allerdings stellt die Suche nach einer geeigneten Benannten Stelle für viele Hersteller aktuell noch ein ernstzunehmendes Problem dar.
In der Summe geht daher der für die Umsetzung der MDR verbundene regulatorische Mehraufwand mit einem erheblich gesteigerten Ressourcenbedarf für die Hersteller einher und generiert Risiken für die medizinische Versorgung durch 1) eine Verkleinerung der Produktpalette, 2) potenzielle Versorgungslücken bei bewährten Medizinprodukten und 3) die potenzielle Behinderung von Innovationen.
5.2 In Vitro Diagnostic Regulation (IVDR)
Die zweite umfassende Neuregelung auf europäischer Ebene ist die In Vitro Diagnostic Regulation (IVDR), die nach fünfjähriger Übergangsfrist im Mai 2022 volle Gültigkeit bekommen sollte. Verschiedene Faktoren haben jedoch hier zu einer schleppenden Umsetzung der IVDR durch europäische Institutionen und Unternehmen geführt. Daher wurde vom EU-Gesetzgeber ein differenzierter, partieller Aufschub der Anwendung um bis zu fünf Jahre beschlossen. Dennoch müssen bereits jetzt alle neu auf den EU-Markt kommenden Diagnostik-Produkte den Vorgaben der IVDR entsprechen.
Durch die massive Steigerung der regulatorischen Anforderungen an die Herstellenden sowohl für die Rezertifizierungen von Bestandsprodukten als auch die Erstzertifizierung von Neuprodukten besteht wie bei der MDR im Bereich der kommerziell verfügbaren industriell hergestellten Diagnostik-Produkte das Risiko, dass diese geänderten Rahmenbedingungen zu einer Mangelsituation etablierter In-vitro-Diagnostik-Tests auf dem Markt führen. In Kombination mit der noch zu geringen Zahl an benannten Stellen und der zum Teil fehlenden Module der EUDAMED-Datenbank kann dadurch in den kommenden Jahren die Versorgung mit essenziellen diagnostischen Tests gefährden werden. Dies gilt insbesondere für den Bereich von Nischen-Untersuchungen mit kleinen Marktsegmenten und Produkte von kleineren Unternehmen. Problematisch in der Handhabung unter IVDR-Bedingungen sind auch die zahlreichen nicht vermarkteten Diagnostik-Artikel aus Eigenherstellung ärztlicher Labore, die je nach Spezialisierungsgrad der Labore bei 50–90% der Untersuchungen zum Einsatz kommen und die vor allem in der personalisierten Medizin unverzichtbar sind oder in frühen Stadien generalisierter Erkrankungen wertvolle diagnostische Informationen liefern. Für die In-vitro-Diagnostika aus Eigenherstellung wird die Interpretation und Umsetzung der IVDR von den Behörden der Bundesländer bestimmt.
5.3 Gesetzgebung zur Nutzung von Registerdaten
Die Problematik der Implementierung von MDR und IVDR zeigt, dass eine transparente und effiziente Umsetzung nicht zuletzt auf der Verfügbarkeit großer Datenmengen in gut organisierten Registern beruht. Medizinische Daten sind dabei nicht nur für die Nachverfolgung und Qualitätskontrolle von Medizinprodukten und Diagnostika essenziell, sondern auch für die medizinische Forschung. Für die medizinische Forschung ist vor allem Zugang zu Daten aus der Versorgung essenziell, um anhand von diesen Daten Hypothesen zu generieren und neue diagnostische und therapeutische Ansätze entwickeln zu können (Sekundärnutzung). Den geregelten, sicheren, datenschutzkonformen und überwachten Zugang zu Daten und Werkzeugen auf Basis eines vertrauenswürdigen Frameworks von Technologien und Regeln zu schaffen, ist seit 2016 das Ziel der Medizininformatik-Initiative (MI-I) des BMBF. Kernelemente der MI-I sind die sogenannte Datenintegrationszentren (DIZ), welche als neue Forschungsinfrastruktur an allen Universitätskliniken aufgebaut und untereinander vernetzt werden. In einem DIZ werden alle Datenspuren aller Patientinnen und Patienten des Standorts in einem klinischen Datenrepositorium zunächst gebündelt, um diese auf Antrag Forschenden zur Verfügung zu stellen. Eine wichtige Grundlage bilden dabei für die DIZ national konsentierte Standards, sowohl für die Daten (z.B. FHIR Profile, Klassifikationen wie ICD-10 und ICD-11, Terminologien wie SNOMED CT) als auch die Prozesse (z.B. Nutzungsordnungen, Nutzungsverträge), was standortübergreifende Projekte wesentlich erleichtert.
Für die Forschungsnutzung werden die zunächst personenbezogenen klinischen Daten unter definierten Bedingungen (u.a. Pseudonymisierung, Broad Consent, zentrales Studienregister) durch eine Treuhandstelle in eine Forschungsdatenrepositorium-Struktur überführt. Dabei entscheiden Use and Access Committees (UAC) anhand eines Datennutzungsantrages, ob methodische, rechtliche und ethische Grundsätze eingehalten werden. Über die DIZ der Universitätsmedizin hinaus sollen die sechs vom BMBF geförderten Digitalen Fortschrittshubs Gesundheit (2021–2025) anhand konkreter medizinischer „Use-Cases“ zeigen, wie die Konzepte der MI-I auch in die regionale Versorgung und intersektorale Forschung übertragen werden können.
Parallel entstehen im Zeitalter der elektronischen Erfassung von Gesundheitsdaten in den Registern der Krankenkassen große Mengen an medizinischen Routinedaten, die für Untersuchungen im Rahmen von Nutzenbewertungen bestimmter Behandlungen oder Therapeutika wertvoll werden können. Weiterhin sieht die kürzlich erlassene Verordnung zum Betrieb eines bundesweiten Implantatregisters die Sammlung umfangreicher Daten zu einer großen Zahl verschiedener Implantattypen vor. In Verbindung mit der bereits erwähnten EUDAMED-Datenbank entstehen hier große Mengen an Daten, die eine wertvolle Ressource für die medizinisch-klinische Forschung darstellen.
Zur Minderung der Risiken der Umsetzung der MDR hat die AWMF für die Fachgesellschaften Konzepte erarbeitet und in drei Stellungnahmen und zwei Anhörungen vorgebracht [18], [19], [20]. Zusammen mit der Deutschen Gesellschaft für Chirurgie fokussiert die AWMF in ihrer Ad-hoc-Kommissionsarbeit aktuell darauf, frühzeitig Versorgungslücken mit Einschränkungen in der Qualität der Patientenversorgung durch Melderegister für Anwender offenzulegen als Basis für Sonderregelungen nach Artikel 59 MDR und weiterführende Überlegungen.
5.4 Empfehlungen
Für die nationale Implementierung der MDR und IVD empfiehlt die AWMF:
- die Vereinfachung der Rezertifizierung von Bestandsprodukten. Hierzu eignet sich als Bewertungskriterium die lange Laufzeit eines Produktes in einem Register ohne Auffälligkeiten unter Definition von Qualitätskriterien. Diese Register sollten über die entsprechenden Fachgesellschaften geführt werden. Die resultierende Entlastung der Benannten Stellen würde wiederum Kapazitäten zur Prüfung von Innovationen freisetzen.
- eine Beschleunigung des Marktzugangs neuer Produkte durch ein „Premarket Approval“ zusammen mit einer kontinuierlichen Datengewinnung, um ein „Rolling Review“ durch die Benannten Stellen zu ermöglichen.
- für Orphan Devices eine Zertifizierung mit Auflagen (z.B. verstärktes PMCF, Aufbau/Nutzung von Produktregistern durch die Fachgesellschaften).
- einen fachlich-wissenschaftlich fundierten Vollzug der IVDR unter Mitwirkung der AWMF Ad-hoc-Kommission In-vitro-Diagnostik. Dies ist notwendig, um vor allem für Innovationen aus der forschenden Hochschulmedizin keine innovationsfeindlichen Hürden aufzubauen.
- eine Vereinheitlichung der Datensammlung in Registern und Repositorien im Kontext der Medizininformatik-Initiative im Hinblick auf Struktur und Terminologie sowie Zugang für eine Nutzung und Auswertung der Daten durch die medizinische Wissenschaft unter Wahrung der gesetzlichen Vorschriften des Datenschutzes.
- die qualitätskontrollierte Kombination der existierenden und entstehenden Datenbanken zur Schaffung von Datenstrukturen, deren wissenschaftliche Evaluierung zur Erweiterung des medizinischen Verständnisses der erfassten Krankheiten beitragen und für entsprechende Konsequenzen für Vorbeugung und Behandlung genutzt werden können.
- die Vorbereitung einer intersektoralen Versorgungstruktur des Gesundheitswesens durch wissenschaftlich gesicherte und evidenzbasierte Daten.
- einen mutigeren Umgang mit den Möglichkeiten des Datenschutzes durch die lokalen Ethikkommissionen auf Basis der „research exemption“ in der Datenschutz-Grundverordnung (DSGVO).
6 Diskussion
Evidenzbasierte Medizin soll die Versorgung von Patientinnen und Patienten steuern. In den letzten drei Jahrzehnten ist dieses Paradigma zunehmend Wirklichkeit geworden, mit einem zusätzlichen Schub in der COVID-19-Pandemie. Voraussetzungen für die Erfüllung eines solchen Anspruchs sind klinische Studien, die so nah wie möglich an der Krankheitsrealität so durchgeführt werden, dass die Ergebnisse rasch in die Versorgung übernommen werden können. Im Berliner Forum 2022 wurden die erreichten Ziele auf der Positivseite und existierende Hürden auf der Negativseite beleuchtet. Letztere müssen in der öffentlichen Diskussion dominieren, wenn Veränderungen stattfinden sollen.
Die aktuellen Entwicklungen bei der Zulassung und Verfügbarkeit von Arzneimitteln und von medizinischen Geräten (Medical Devices) illustrieren einige Kernprobleme der bidirektionalen Interaktion zwischen Krankenversorgung und Schaffung von wissenschaftlicher Evidenz (Tabelle 3 [Tab. 3]).
Tabelle 3: Kernforderungen der AWMF zum besseren Transfer der Evidenz in die Versorgung
Bürokratische Hürden 1
Die gut gemeinte EU Clinical Trials Regulation setzt hohe bürokratische Hürden für akademische Entwickler und andere kleine Institutionen. Netzwerke wie das KKS bieten Unterstützung, werden aber in Deutschland durch zusätzliche nationale Regelungen gebremst. Manch große internationale Studien hat die Rekrutierungsphase bereits beendet, bevor alle Hürden in Deutschland überwunden sind. Die EU hat die Probleme wahrgenommen und die Netzwerkstrategie 2025 entwickelt. Auch in Deutschland müssen überflüssige Hürden beseitigt und Abläufe durch enge Fristen beschleunigt werden.
Bürokratische Hürden 2
Die gut gemeinte Medical Device Regulation erweist sich ebenfalls als hinderlich, einerseits für die Anmeldung von Innovationen durch akademische Einrichtungen und kleine und mittlere Unternehmen, andererseits für die Verfügbarkeit von Bestandsprodukten durch deren (Re-)Registrierungsaufwand.
Kommerzielle Interessen
Gerade der Bereich der Arzneimittel und der Medical Devices wird von kommerziellen Interessen global agierender Firmen dominiert. Auf der positiven Seite gäbe es die meisten Fortschritte ohne das Engagement der Industrie nicht. Entsprechend hoch sind die Erwartungen der Firmeninhaber und der Shareholder nach einer Zulassung auf dem deutschen Markt. Für die wissenschaftlichen medizinischen Fachgesellschaften ist die Wahrung der geistigen und der wirtschaftlichen Unabhängigkeit eine große Herausforderung. Maximale Transparenz bei möglichen Interessenkonflikten ist eine unverzichtbare Voraussetzung.
Unabhängige Studien
Die starke wirtschaftliche Kraft der Industrie hat eine noch in den 1980er Jahren bestehende Kultur großer, von Ministerien finanzierter, multizentrischer Studien fast vollständig verdrängt. Diese unabhängige, an der Versorgung in Deutschland orientierte Studienkultur muss wieder aufgebaut werden.
Health Technology Assessment versus evidenzbasierte Medizin
Dieser Antagonismus ist kein grundsätzliches Gegensatzpaar; viele methodische Aspekte überlappen. Dennoch ist der Ansatz Health Technology Assessment mit der jetzt gerade wieder beginnenden Bewertung von Arzneimitteln/Methoden auf der Basis von QALYs (qualitätsadjustierte Lebensjahre) eine intentionell getriebene Bewertung mit dem Ziel der Kostendämpfung. Nicht unerwartet weichen Empfehlungen von Leitlinien von HTA-Bewertungen substanziell ab. Die Leitlinien sind nahe an der Versorgung von Patientinnen und Patienten. Sie sind aber nur so gut wie die Qualität ihrer Datenbasis, der Prozess ihrer Ausarbeitung und ihre Aktualität. Diese Herausforderung kollidiert oft mit dem ehrenamtlichen Engagement der Erstellenden von Leitlinien.
7 Fazit
Das Berliner Forum hat das hohe Niveau der Krankenversorgung in Deutschland unterstrichen, inkl. der zeitnahen Versorgung mit Arzneimitteln und mit Medical Devices. Es hat aber auch sehr deutlich gemacht, dass der Erhalt des Erreichten und weiterer Fortschritt Veränderungen in der Gewinnung von Evidenz im Rahmen klinischer Studien und in der Bewertung der Ergebnisse erfordern. Dabei sollten medizinisch begründeter Bedarf einerseits und die Sicherheit von Patientinnen und Patienten andererseits im Vordergrund stehen. Die in diesem Positionspapier der AWMF hergeleiteten Forderungen sind realistisch, konsensfähig und sollten zügig umgesetzt werden.
Anmerkungen
Danksagungen
Wir danken den externen Vortragenden beim Berliner Forum Karl Broich, Ulrich M. Gassner, Josef Hecken, Britta Lang und allen an den Diskussionen beteiligten Anwesenden für ihre Anregungen zu diesem Artikel.
Interessenkonflikte
Die Autorinnen und Autoren erklären, dass sie keine Interessenkonflikte in Zusammenhang mit diesem Artikel haben.
Literatur
[1] Sachverständigenrat für die Konzertierte Aktion im Gesundheitswesen. Gesundheitsversorgung und Krankenversicherung 2000. Mehr Ergebnisorientierung, mehr Qualität und mehr Wirtschaftlichkeit. Kurzfassung und Empfehlungen, Sondergutachten 1995. Baden-Baden: 1995. Available from: https://www.svr-gesundheit.de/gutachten/sachstandsbericht-1995/[2] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Positionen der AWMF zur Gesundheitspolitik nach der Bundestagswahl 2017. Evidenzbasierte Medizin - die Basis einer guten Gesundheitspolitik. Version 1.0. Berlin: AWMF; 2017 Jun 16. Available from: https://www.verwaltung.awmf.org/fileadmin/user_upload/Stellungnahmen/Resolution_Forderungen/AWMF_Position_Gesundheitspolitik_lang_2017-06.pdf
[3] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Positionen der AWMF zur Gesundheitspolitik nach der Bundestagswahl 2021. Evidenzbasierte Medizin - die Basis einer guten Gesundheitspolitik. Version 3.2. Berlin: AWMF; 2021 Mar 18. Available from: https://www.verwaltung.awmf.org/fileadmin/user_upload/Stellungnahmen/Resolution_Forderungen/20210318_Master_Gesundheitspolitik_2021_AWMF_Position_3.2_lang_fin.pdf
[4] Treede RD. Forschungsförderung: Status und zukünftige Herausforderungen aus Sicht der AWMF. Frankfurter Forum: Diskurse. 2023 Jan;2023(26):6-13.
[5] European Medicines Agency. Clinical Trials Regulation. [last accessed 2023 Aug 14]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trials-regulation
[6] Deutsche Gesellschaft für Hämatologie und medizinische Onkologie. Frühe Nutzenbewertung neuer Arzneimittel in Deutschland - Subgruppen Definition, Analyse und Kriterienkatalog. In Kooperation mit der Ad-Hoc-Kommission Nutzenbewertung der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF). Berlin: DGHO; 2016. (Gesundheitspolitische Schriftenreihe der DGHO; 8). Available from: https://www.dgho.de/publikationen/schriftenreihen/fruehe-nutzenbewertung
[7] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF); Deutsche Gesellschaft für Hämatologie und medizinische Onkologie; Ad-hoc-Kommission „Nutzenbewertung von Arzneimitteln“. Frühe Nutzenbewertung neuer Arzneimittel in Deutschland 2011-2018 – Gerechtigkeit und Nachhaltigkeit. Berlin: AWMF/DGHO; 2019 May. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/nutzenbewertungen/AWMF_AMNOG_2019_210x297_F_Einzelseiten.pdf
[8] Netzwerk Koordinierungszentren Klinischer Studien (KKS-Netzwerk) e.V. [last accessed 2023 Aug 14]. Available from: https://www.kks-netzwerk.de/
[9] Nothacker M, Muche-Borowski C, Kopp IB. 20 Jahre ärztliche Leitlinien in Deutschland - was haben sie bewirkt? [Reflections on 20 years of clinical practice guideline programmes in Germany: what is their impact?]. Z Evid Fortbild Qual Gesundhwes. 2014;108(10):550-9. DOI: 10.1016/j.zefq.2014.10.012
[10] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). AWMF-Leitlinienregister. [last accessed 2023 Aug 14]. Available from: https://register.awmf.org/de/start
[11] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Von der Planung bis zur Publikation: AWMF-Regelwerk Leitlinien. Version 2.1 vom 05.09.2023. [last accessed 2023 Aug 14]. Available from: https://www.awmf.org/regelwerk/
[12] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Informationen und Leitlinien: COVID-19-Pandemie. [last accessed 2023 Aug 14]. Available from: https://www.awmf.org/aktuelle-leitlinien-und-informationen-zu-covid-19
[13] Covid-19 Evidenz-Ökosystem (CEO-sys). [last accessed 2023 Aug 14]. Available from: https://covid-evidenz.de/
[14] Hecken J. Förderung durch den G-BA: Entwicklung und Implementierung hochwertiger Leitlinien zur Verbesserung der Versorgung. In: Berliner Forum 2022; 2022 Apr 27; Berlin. [last accessed 2023 Aug 14]. Available from: https://www.awmf.org/die-awmf/veranstaltungen/berliner-forum-der-awmf
[15] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Digitalisierung von Leitlinienwissen - Gesamtkonzept der AWMF. [last accessed 2023 Aug 14]. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/medizinische_versorgung/it-projekt-gesamtkonzept-awmf-abbildung.pdf
[16] Kopp I, Nothacker M, Spies C, Müller A, Gamstätter T, Langer T, Wenzel G, Starlinger J, Karge T, Vandvik P, Brandt L. Modellprojekt für die Digitalisierung hochwertiger Leitlinien und deren Verbreitung über Apps (DissolVe). Marburg/Berlin: AWMF; 2020. Available from: https://www.bundesgesundheitsministerium.de/service/publikationen/gesundheit/details.html?bmg%5Bpubid%5D=3583
[17] MAGIC Evidence Ecosystem Foundation. MAGIC. [last accessed 2023 Aug 14]. Available from: https://app.magicapp.org/
[18] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Stellungnahme zum Implantatverbleib der AWMF-ad hoc Kommission “Bewertung von Medizinprodukten“. Berlin: AWMF; 2018. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/stellungnahmen/2018/20180622_Stellungnahme_Umgang_mit_Explantaten_fin.pdf
[19] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Stellungnahme der Ad hoc Kommission Nutzenbewertung von Medizinprodukten der AWMF zum Entwurf einer Verordnung zum Betrieb des Implantateregisters Deutschland (Implantateregister-Betriebsverordnung -IRegBV). Berlin: AWMF; 2021 Jul 15. Available from: https://www.verwaltung.awmf.org/fileadmin/user_upload/Stellungnahmen/Medizinische_Versorgung/20210715_AWMF_SN_IRegBV_final.pdf
[20] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Positionspapier der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF e.V.) zur Implementierung der Medical Device Regulation (MDR). Berlin: AWMF; 2022 Dec 12. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/stellungnahmen/2022/20221218_Implementierung_der_Medical_Device_Regulation__MDR_.pdf
[21] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Langfassung zu der Position der AWMF zur Gesundheitspolitik nach der Bundestagswahl 2017: Evidenzbasierten Medizin – die Basis einer guten Gesundheitspolitik vom 16.06.2017. Version 1.0. Berlin: AWMF; 2017 Jun 16. Available from: https://www.awmf.org/die-awmf/awmf-stellungnahmen/langfassung-zu-der-position-der-awmf-zur-gesundheitspolitik-nach-der-bundestagswahl-2017-evidenzbasierten-medizin-die-basis-einer-guten-gesundheitspolitik-vom-16062017
[22] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Position der AWMF zur Gesundheitspolitik nach der Bundestagswahl 2021 – Evidenzbasierte Medizin – die Basis einer guten Gesundheitspolitik (Langfassung). Version 3.2. Berlin: AWMF; 2021 Mar 18. Available from: https://www.awmf.org/die-awmf/awmf-stellungnahmen/position-der-awmf-zur-gesundheitspolitik-nach-der-bundestagswahl-2021-evidenzbasierte-medizin-die-basis-einer-guten-gesundheitspolitik-langfassung
[23] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Berliner Forum. Available from: https://www.awmf.org/die-awmf/veranstaltungen/berliner-forum-der-awmf
[24] Heads of Medicines Agencies; European Commission; European Medicines Agency. Accelerating Clinical Trials in the EU (ACT EU) – Delivering an EU clinical transformation initiative. 2022 Jan 13. Available from: https://www.ema.europa.eu/en/news/accelerating-clinical-trials-eu-act-eu-better-clinical-trials-address-patients-needs
[25] European Medicines Agency. Annual Report 2022 – The European Medicines Agency’s contribution to science, medicines and health in 2022. Amsterdam: Publications Office of the European Union; 2023. DOI: 10.2809/08948
[26] European Commission: Directorate-General for Health and Food Safety. Study on an infrastructure and data ecosystem supporting the impact assessment of the European health data space – Executive summary. Publications Office of the European Union; 2022. DOI: 10.2875/406794

