Chronic central serous choroidopathy with severe visual loss in hyperopic microphthalmic identical twins
Annelies Fabry 1Johan Blanckaert 1
Anita Leys 1
1 Department of Ophthalmology, University Hospitals Leuven, Leuven, Belgium
Abstract
Objective: To report chronic central serous choroidopathy with severe visual loss in hyperopic microphthalmic identical twins.
Methods: The index patient was first examined in 1994, at age 31, and has been followed up closely for 17 years. He had repeated fluorescein and indocyanine green angiograms, OCT, ultrasound biometry, and recently also autofluorescence and EDI OCT. His twin brother was first examined in 2010, at age 47, and had a similar extensive exploration.
Results: The index patient had a mean refractive error of +6 D OU and VA was 20/32++ in the RE and 20/200 in the LE in 1994. Vision slowly went down to 20/800+ in the RE and 20/600 in the LE in 2011. His twin brother has a mean refractive error of +6 D OU and VA 20/400 OU. Both have a short axial length of the eye, a thick choroid with dilated vessels, and central serous choroidopathy with cystic degeneration of the macula and retina in the posterior pole.
Conclusions: We add to the reported complications of microphthalmos, chronic central serous choroidopathy.
Introduction
So far, hyperopia is not considered to be a risk factor for central serous choroidopathy (CSC). Twins with high hyperopia were reported by Black in 1924 [1] and CSC has been observed in identical twins by Jonkers in 1960 [2], but none had the combined pathology. Familial CSC [3] and familial chronic CSC [4] without hyperopia have been reported by others. We report CSC with severe visual loss in hyperopic identical twins. Ultrasound A-scan biometry showed reduced total total axial lenght of the globes, and EDI OCT showed dilated choroidal veins and a thick choroid. These findings show that hyperopia with short axial length of the eye can predispose to CSC [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23].
Case presentation
Index patient
The index patient started wearing spectacles at age 14, when he was learning weaving techniques. For several years the hyperopic correction was increased, and the corrected VA allowed the patient to work easily. At age 27, vision loss was noticed, first in the left eye and later in the right eye. His ophthalmologist was unable to correct the vision, diagnosed a maculopathy, and referred the patient to a medical retina specialist, who advised lasercoagulation. The patient did not want this treatment, waited for some time, and came to see us for second opinion in 1994, at age 31.
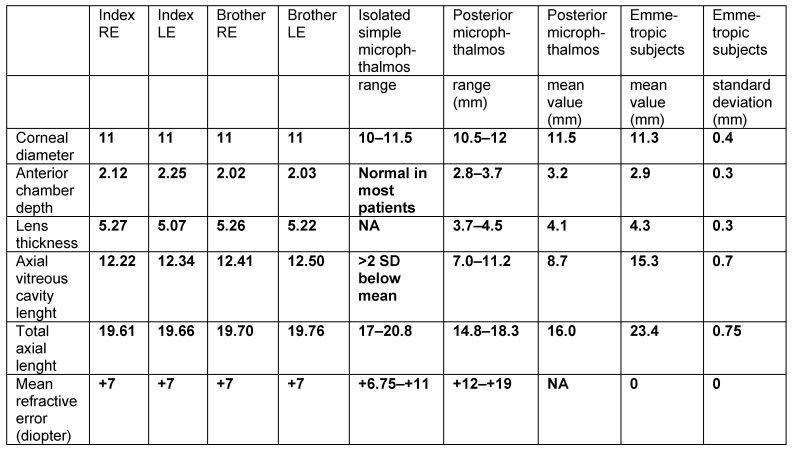
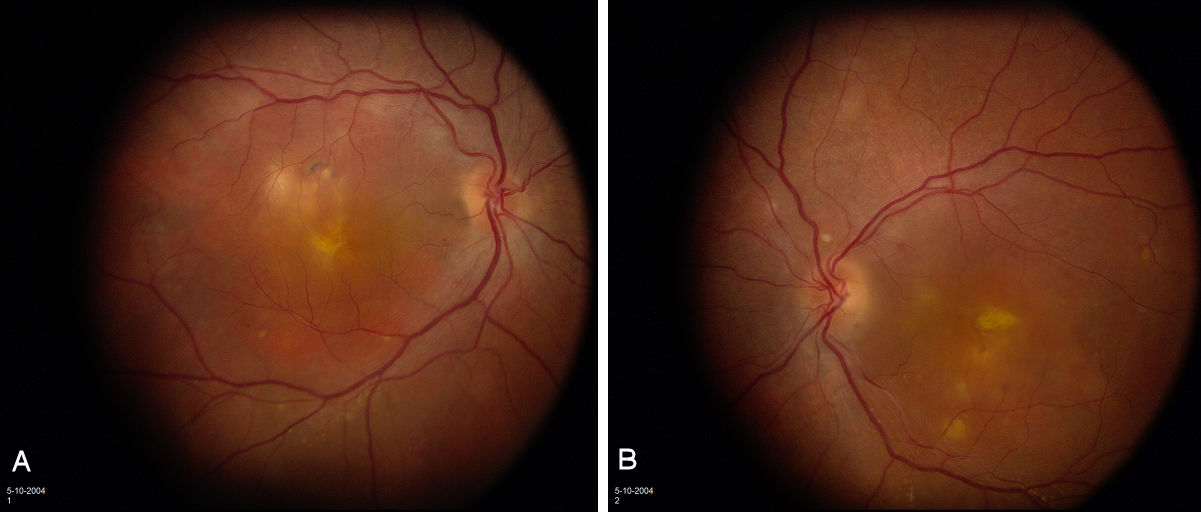
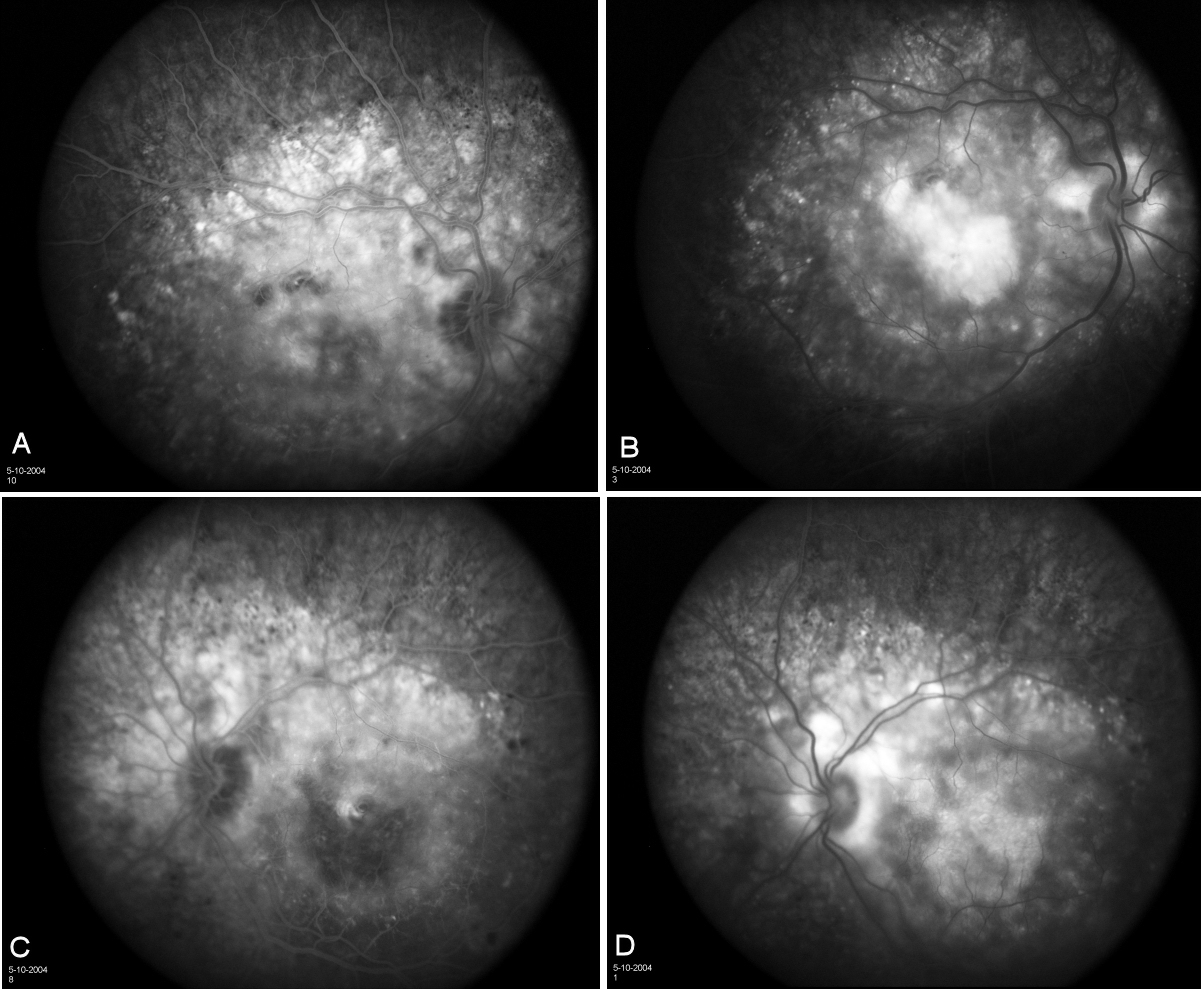
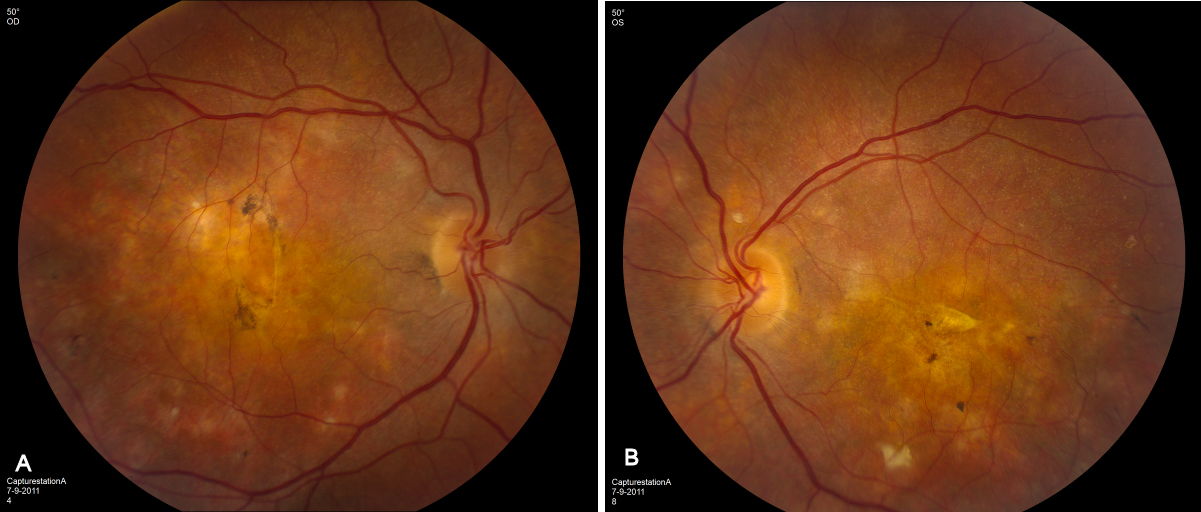
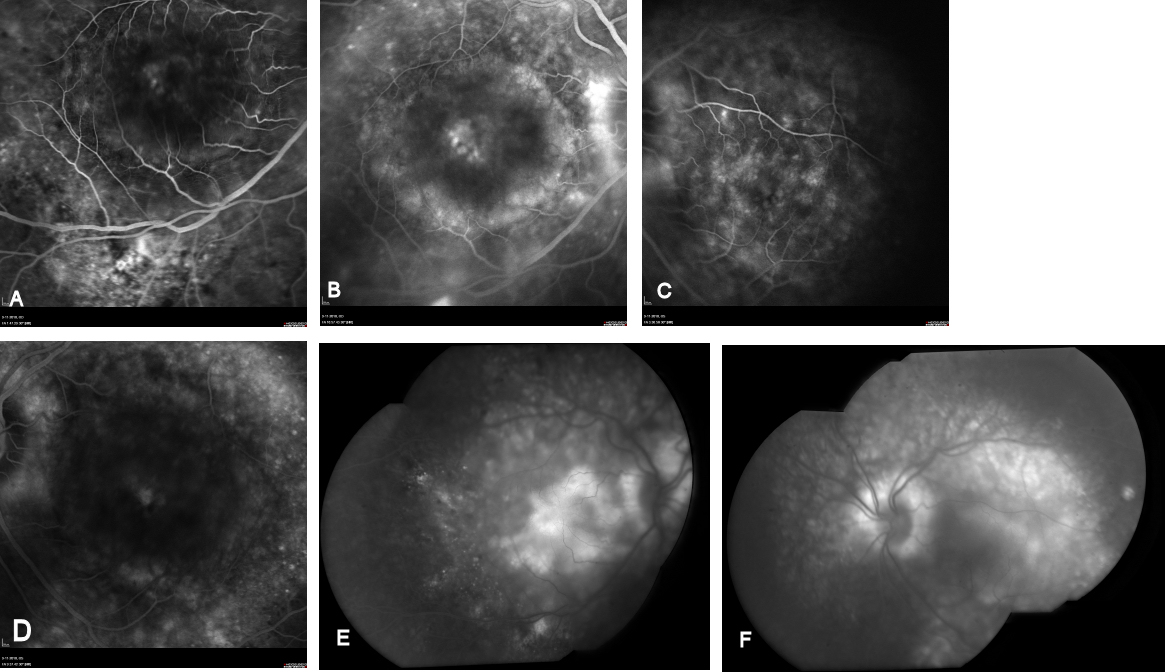
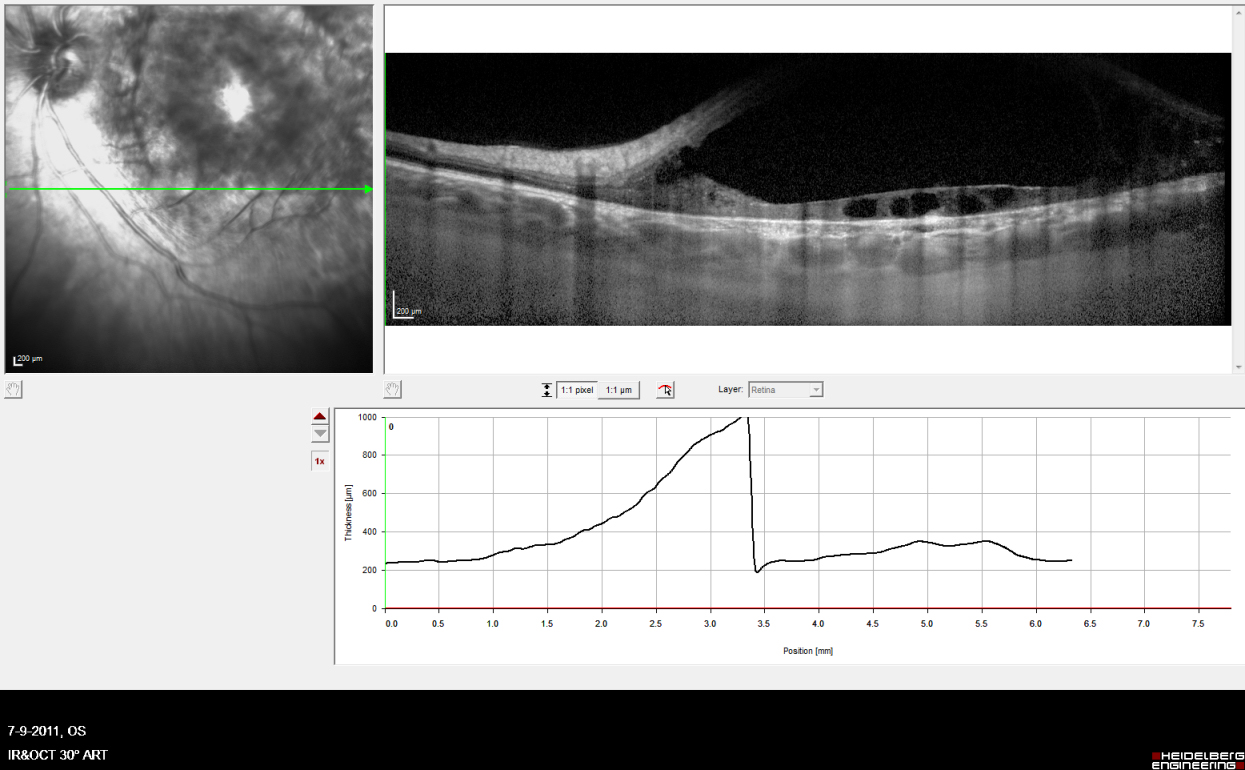
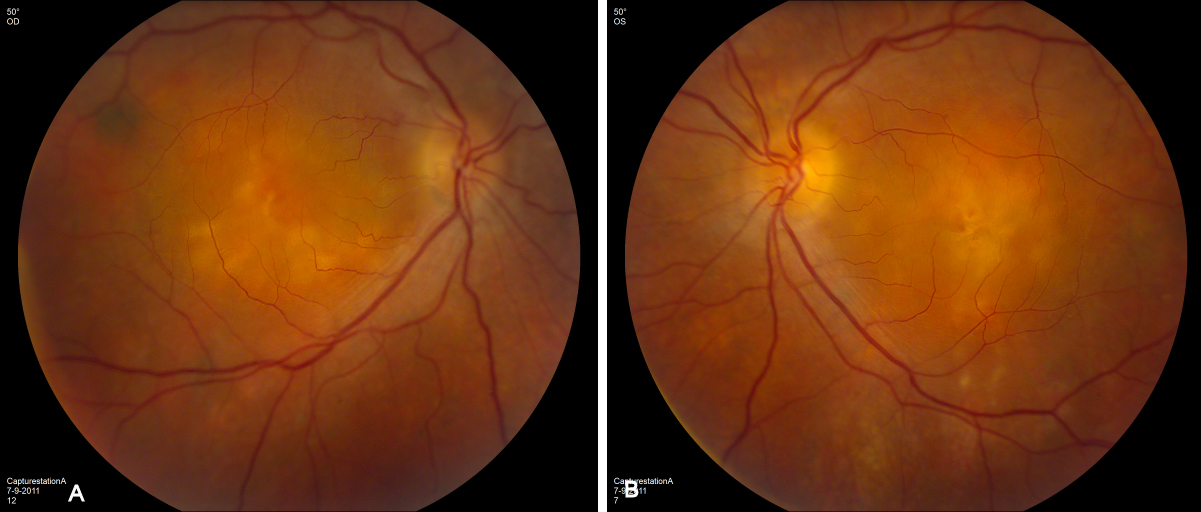
External examination of the eyes was normal, including corneal size (11 mm diameter OU) and palpebral fissure (Figure 1 [Fig. 1]). BCVA was 20/32++ in the RE and 20/200 in the LE (correction RE sph +5.25 cyl +1.50 axis 42 and LE +5.50 cyl +0.75 axis 109). Ocular tension was 17 mmHg RE and 18 mmHg LE. Biomicroscopic examination showed in both eyes a mildly reduced anterior chamber depth and a maculopathy (Figure 2 [Fig. 2]). Fluorescein angiography confirmed the presumed diagnosis of CSC OU with macular serous detachment, diffuse pigment epitheliopathy, and mild focal subretinal leakage (Figure 3 [Fig. 3]). Focal lasercoagulation was performed in the RE in 1995, 1996, 1997 and 1998, but despite this treatment the macular detachment increased slowly and cystic retinal degeneration and fibrosis became evident, as well in the RE as in the LE (Figure 4 [Fig. 4]). Repeated FA revealed progressively expanding subretinal lesions in the posterior pole of both eyes, with leakage, staining and pooling of fluid (Figure 5 [Fig. 5]). In 2004, PDT was available, and as the exudative lesions were pronounced, one session of SFR PDT was administered in the LE, however, without benefit. Over the last years of follow-up eye drops with dorzolamide were used, apparently without benefit. OCT was available in 1997, and repeated scans showed pronounced and progressive cystic degeneration of the macula OU (Figure 6 [Fig. 6]). EDI OCT, available in 2011, demonstrated dilated choroidal vessels and a thick choroid (Figure 7 [Fig. 7]). ICGA also showed dilated choroidal vessels (Figure 8 [Fig. 8]). Ultrasound A-scan biometry demonstrated OU reduced total axial length, reduced anterior chamber depth, reduced vitreous cavity and increased lens thickness (Table 1 [Tab. 1]). Recently, atrophic changes became evident (Figure 9 [Fig. 9]) and on OCT decreased retinal thickness was observed (Figure 7 [Fig. 7]). Autofluorescence imaging showed large areas of hypoautofluorescence compatible with afunctional retinal pigment epithelium and retina (Figure 10 [Fig. 10]). VA in 2011 was 20/800+ in the RE and 20/600 in the LE.


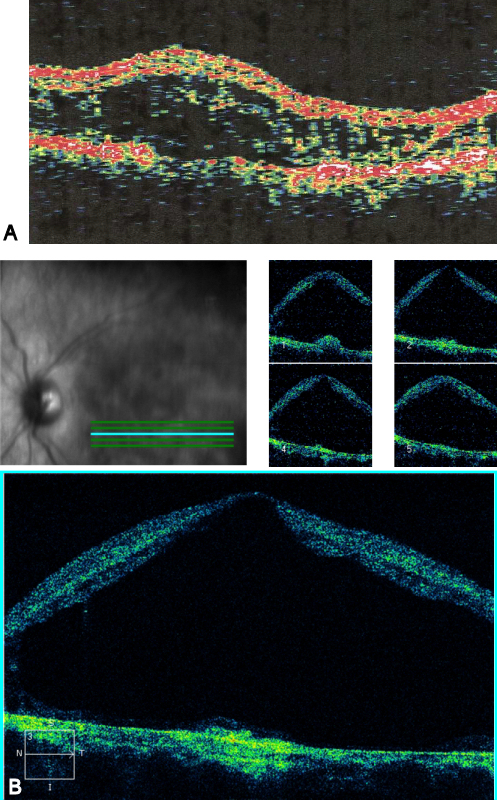


Twin brother
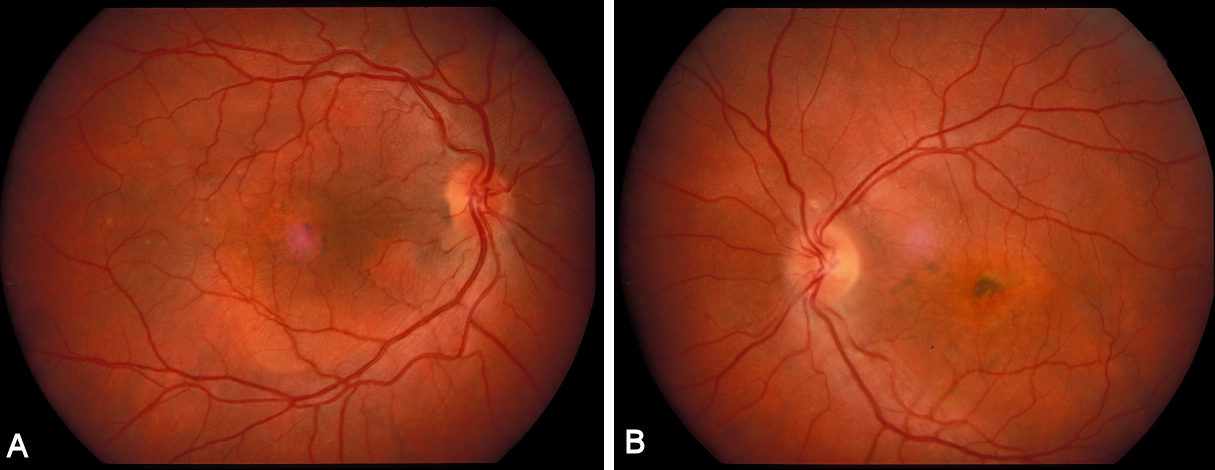
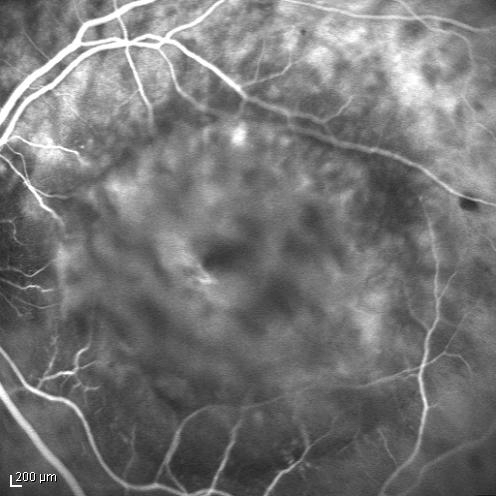
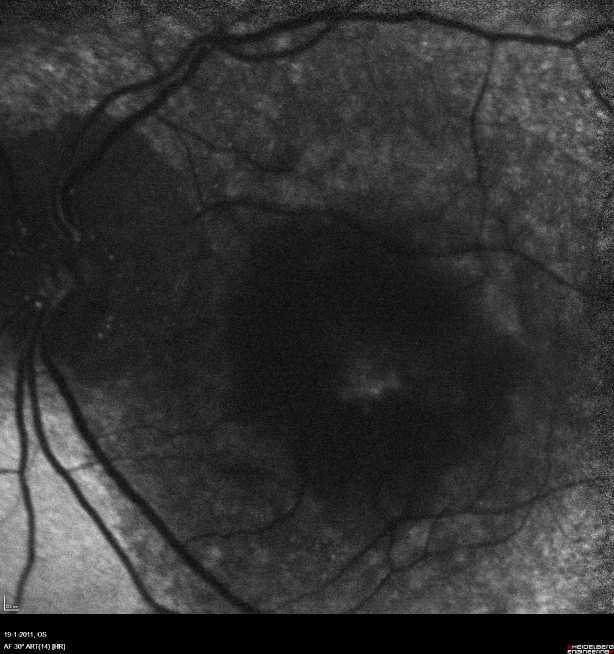
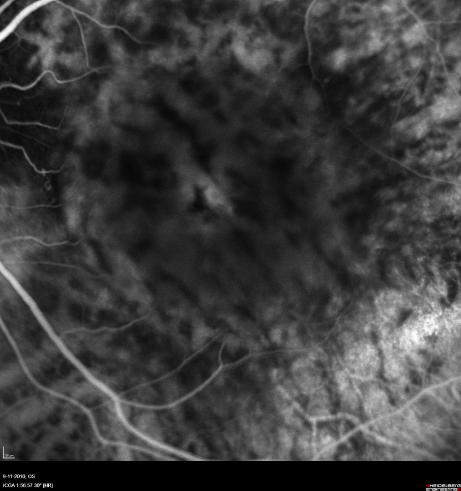
The twin brother received his first pair of glazes at age 21. He is working as a gardener without major visual problems, but at age 34 developed reading problems and vision loss. At age 47, he realized to have a similar eye problem as his twin brother, and consulted for diagnosis and testing of low vision aids. External examination of the eyes was normal, including corneal size (diameter 11 mm OU) and palpebral fissures (Figure 1 [Fig. 1]). BCVA was 20/400 OU (RE sph +6.25 cyl –0.75 axis 113° and LE sph 7.00 cyl –1.75 axis 67°). Ocular pressure was normal (16 and 15 mmHg on pneumotonometry). Biomicroscopic examination showed a mildly reduced anterior chamber depth and a bilateral maculopathy with CSC very similar to his brothers. Autofluorescence imaging (Figure 11 [Fig. 11]), fluorescein angiography (Figure 12 [Fig. 12]) and indocyanine green angiography (Figure 13 [Fig. 13]), OCT and EDI-OCT (Figure 14 [Fig. 14]), and ultrasound biometry (Table 1 [Tab. 1]) were performed and showed similar ocular anomalies and a maculopathy that was slightly milder than his brother’s. Over the next year the maculopathy got slightly worse (Figure 15 [Fig. 15]).
Discussion
The twins reported in this paper are 6 diopters hyperopic in both eyes, have normal corneal size, and presented with bilateral chronic central serous choroidopathy and severe visual loss. The four eyes have similar biometric ocular data with mildly reduced anterior chamber depth, a lens thicker than expected, reduced vitreous cavity and reduced total axial length. These data confirm a developmental anomaly in the spectrum of microphthalmos [24] (Table 1 [Tab. 1]).
Microphthalmos is a congenital ocular malformation with reduced size of the globe. The clinical suspicion can be raised on the basis of apparently small eyes [25]. In suspected patients another condition, blepharophimosis, should be excluded. Blepharophimosis is characterized by small palpebral fissures, but normal size of the globe [25].
Microphthalmos can present as an isolated ocular anomaly (isolated simple or pure microphthalmos), or in association with other ocular or systemic abnormalities. The spectrum of associated anomalies is broad, and includes coloboma, mental retardation, neurologic and renal anomalies, fetal alcohol syndrome, myotonic dystrophy and achondroplasia [9], [26], [27]. Both autosomal dominant and recessive forms of inheritance have been reported in microphthalmos. According to Warburg [25], [26], there are more than 130 syndromes which may present microphthalmos and over 70 with blepharophimosis, but in only 13 cases are the two found in the same syndrome. So far, the molecular genetics of the morphogenesis of the eye and, thus of microphthalmos, have revealed that PAX6 at 11p13, PAX3 at 2q35, and the microphthalmia transcription factor MITF are of importance for the embryonic growth of the eye, but blepharophimosis has not been reported in patients with mutations or deletions of these genes [25]. We have reported microphthalmos in a patient with renal-coloboma syndrome associated with PAX2 mutation [27]. Sundin et al. [19] have shown that recessive nanophthalmos is caused by severe mutations in the MFRP gene at 11q23.3. This gene encodes the Frizzled-related transmembrane protein MFRP that is selectively expressed in the retinal pigment epithelium (RPE) and ciliary body. MFRP protein is required for both prenatal ocular growth and postnatal emmetropization. Othman et al., reported autosomal dominant nanophthalmos (NNO1) with high hyperopia and angle-closure glaucoma mapping to chromosome 11 [14].
Whenever a patient is suspected of having microphthalmos, an ultrasound determination of the axial length of the eye should be provided [25]. Ultrasound biometry allows accurate assessment of the total axial length of the globe, the anterior chamber depth and risk for angle closure glaucoma [14], [18], and the volume of lens and vitreous cavity. Based on biometric data harmonious microphthalmos is differentiated of posterior microphthalmos. Microphthamos can be extreme with blindness [27] or relatively mild with spared vision [9].
Posterior microphthalmos disproportionately affects the posterior ocular segment with normal external appearance of the eyes [6]. The corneal size and the anterior segment dimensions are normal, but there is foreshortening of the posterior ocular segment and high hyperopia. This hyperopia and an elevated papillomacular retinal fold are the main causes of visual impairment [6], [12], [13], [16], [17], [21]. A disparity in growth between the sclera and retina probably gives rise to the frequently identified elevated papillomacular retinal fold [6], [16].
Other relatively frequent ocular abnormalities observed in microphthalmos/posterior microphthalmos are absence of the capillary-free zone, retinal striae, chorioretinal folds, uveal effusion, pigmentary retinal dystrophy and pseudopapilledema [13], [16], [17]. Macular hole, retinitis punctate albescens, and acquired retinoschisis [20] have been reported as a rare association.
The terminology of nanophthalmos has been used mainly in poorly defined phenotypes with small eyes, thick sclera and uveal effusion [5], [7], [8], [15], [23]. The thickened sclera impedes vortex venous drainage and/or reduces transscleral flow of protein from the eye, which causes fluid accumulation (from the choriocapillaris) in the choroid, subretinal space, or both. This may produce choroidal congestion, choroidal detachment, and/or exudative retinal detachment.
The twins reported in this paper have biometric data with reduced total axial length. Their data (Table 1 [Tab. 1]) only mildly differ from those reported by Weiss et al. in 1989 in a study of 10 patients with isolated simple microphthalmos [9] and from those reported by Khairallah et al. in 2002 in a study of 18 patients with posterior microphthalmos [16]. We conclude that high hyperopia urges the ophthalmologist to perform ultrasound biometry to confirm or exclude microphthalmos. Eyes with reduced axial length are microphthalmic and require additional investigation to detect associated anomalies and follow up should be proposed as there is a risk of treatable complications. A shallow anterior angle implies a risk of angle closure glaucoma [7], [18], a treatable condition. Uveal effusion is another treatable complication of microphthalmos [8]. We add to the reported complications of microphthalmos, chronic central serous choroidopathy. Although we failed to stabilize vision, new therapies may become available in the next future making this complication treatable.
Notes
The authors have no notes/conflict of interest concerning the report of this case.
References
[1] Black M. Twins with high hyperopia. Am J Ophthalmol. 1924;7:375-6.[2] Jonkers GH. Central serous retinopathy in a monozygotic twin-pair. Acta Genet Metabol Gemelli. 1960;IX:438-41.
[3] Lin E, Arrigg PG, Kim RY. Familial central serous choroidopathy. Graefes Arch Clin Exp Ophthalmol. 2000;238:930-1. DOI: 10.1007/s004179900110
[4] Weeninck AC, Borsje RA, Oosterhuis JA, et al. Familial chronic central serous chorioretinopathy. Ophthalmologica. 2001;215:183-7. DOI: 10.1159/000050855
[5] Brockhurst RJ. Nanophthalmos with uveal effusion. A new clinical entity. Arch Ophthalmol. 1975;93(12):1989-99.
[6] Spitznas M, Gerke E, Bateman JB. Hereditary posterior microphthalmos with papillomacular fold and high hyperopia. Arch Ophthalmol. 1983;101(3):413-7.
[7] Ghose S, Sachdev MS, Kumar H. Bilateral nanophthalmos, pigmentary retinal dystrophy, and angle closure glaucoma – a new syndrome? Br J Ophthalmol. 1985;69:624-8. DOI: 10.1136/bjo.69.8.624
[8] Allen KM, Meyers SM, Zegarra H. Nanophthalmic uveal effusion. Retina. 1988;8(2):145-7. DOI: 10.1097/00006982-198808020-00012
[9] Weiss AH, Kousseff BG, Ross EA, Longbottom J. Simple microphthalmos. Arch Ophthalmol. 1989;107(11):1625-30.
[10] Vingolo EM, Steindl K, Forte R, Zompatori L, Iannaccone A, Sciarra A, Del Porto G, Pannarale MR. Autosomal dominant simple microphthalmos. J Med Genet. 1994;31(9):721-5. DOI: 10.1136/jmg.31.9.721
[11] Neelakantan A, Venkataramakrishnan P, Rao BS, Krishnan N, Vijaya L, John S, Kar B. Familial nanophthalmos: management and complications. Indian J Ophthalmol. 1994;42(3):139-43.
[12] Kida Y, Kurome H, Hayasaka S. Bilateral microphthalmos with poor visual acuity, high hyperopia, and papillomacular retinal folds in siblings. Jpn J Ophthalmol. 1995;39(2):177-9.
[13] Serrano JC, Hodgkins PR, Taylor DS, Gole GA, Kriss A. The nanophthalmic macula. Br J Ophthalmol. 1998;82(3):276-9. DOI: 10.1136/bjo.82.3.276
[14] Othman MI, Sullivan SA, Skuta GL, Cockrell DA, Stringham HM, Downs CA, Fornés A, Mick A, Boehnke M, Vollrath D, Richards JE. Autosomal dominant nanophthalmos (NNO1) with high hyperopia and angle-closure glaucoma maps to chromosome 11. Am J Hum Genet. 1998;63(5):1411-8. DOI: 10.1086/302113
[15] Yamani A, Wood I, Sugino I, Wanner M, Zarbin MA. Abnormal collagen fibrils in nanophthalmos: a clinical and histologic study. Am J Ophthalmol. 1999;127(1):106-8. DOI: 10.1016/S0002-9394(98)00302-X
[16] Khairallah M, Messaoud R, Zaouali S, et al. Posterior segment changes associated with posterior microphthalmos. Ophthalmology. 2002;109:569-74. DOI: 10.1016/S0161-6420(01)00996-4
[17] Khan AO. Recognizing posterior microphthalmos. Ophthalmology. 2006;113:718. DOI: 10.1016/j.ophtha.2006.01.013
[18] Bron A. Surgical indications in nanophthalmos. J Fr Ophtalmol. 2007;30(2):186-8. DOI: 10.1016/S0181-5512(07)89574-X
[19] Sundin OH, Dharmaraj S, Bhutto IA, Hasegawa T, McLeod DS, Merges CA, Silval ED, Maumenee IH, Lutty GA. Developmental basis of nanophthalmos: MFRP Is required for both prenatal ocular growth and postnatal emmetropization. Ophthalmic Genet. 2008;29(1):1-9. DOI: 10.1080/13816810701651241
[20] Dhrami-Gavazi E, Schiff WM, Barile GR. Nanophthalmos and acquired retinoschisis. Am J Ophthalmol. 2009;147(1):108-10. DOI: 10.1016/j.ajo.2008.07.045
[21] Khan AO. Posterior microphthalmos versus nanophthalmos. Ophthalmic Genet. 2008;29(4):189. DOI: 10.1080/13816810802258862
[22] Khan A, Zafar SN. Variable retinal presentations in nanophthalmos. J Pak Med Assoc. 2009;59(11):791-3.
[23] Moradian S, Kanani A, Esfandiari H. Nanophthalmos. Journal of ophthalmic and vision research. 2011;6(2):145-6. Available from: http://www.jovr.ir/index.php/jovr/article/viewFile/298/323
[24] Delmarcelle Y, François J, Goes F, et al. Biométrie oculaire clinique (oculométrie). Bull Soc belge Ophthalmol. 1976;172:1-331.
[25] Warburg M. Letter to the editor. Small eyes: descriptional misconceptions. Am J Med Genetics. 1995;59:388-9. DOI: 10.1002/ajmg.1320590322
[26] Warburg M. Genetics of microphthalmos. Int Opthalmol. 1981;4:45-65. DOI: 10.1007/BF00139580
[27] Schimmenti LA, Cunliffe HE, McNoe LA, Ward TA, et al. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am J Hum Genet. 1997;60(4):869-78.

